
Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
Usher syndrom
Medicinsk expert av artikeln

Senast recenserade: 04.07.2025
Usher syndrom är en ärftlig sjukdom som yttrar sig som fullständig dövhet från födseln, samt progressiv blindhet med åldern. Synförlust är förknippad med retinitis pigmentosa, en process med pigmentdegeneration av näthinnan. Många personer med Usher syndrom har också allvarliga balansproblem.
Epidemiologi
Tack vare forskningen kunde man fastställa att Usher syndrom drabbar cirka 8 % av de undersökta dövstumma barnen (tester utfördes på särskilda institutioner för dövstumma personer). Pigmentär retinit observerades hos 6–10 % av patienter som lider av medfödd dövhet, vilket i sin tur observeras hos cirka 30 % av personer med pigmentär näthinnesjukdom.
Man tror att denna sjukdom uppträder hos ungefär 3–10 personer av 100 000 människor världen över. Den kan observeras hos både kvinnor och män i lika hög grad. Cirka 5–6 % av världens befolkning lider av detta syndrom. Cirka 10 % av alla fall av djup dövhet i barndomen beror på Usher syndrom I, såväl som typer II.
I USA är typ 1 och 2 de vanligaste typerna. Tillsammans står de för cirka 90 till 95 procent av alla fall av Usher syndrom hos barn.
Orsaker Usher syndrom
Usher syndrom typ I, II och III har en autosomalt recessiv orsak, medan typ IV anses vara en X-kromosomrubbning. Orsakerna till blindhet och dövhet som uppstår vid detta syndrom har ännu inte studerats tillräckligt. Det antas att personer med denna sjukdom är överkänsliga mot komponenter som kan skada DNA-strukturen. Dessutom kan denna sjukdom vara förknippad med immunförsvarsstörningar, men i detta fall finns det ingen exakt bild av denna process.
År 1989 identifierades kromosomavvikelser för första gången hos patienter med typ II-sjukdom, vilket i framtiden kan leda till ett sätt att isolera de gener som orsakar syndromet. Det kan också vara möjligt att identifiera dessa gener hos bärare och utveckla speciella prenatala genetiska tester.
 [ 8 ]
[ 8 ]
Riskfaktorer
Syndromet ärvs när båda föräldrarna är drabbade, dvs. det ärvs av en recessiv typ. Ett barn kan också ärva sjukdomen om föräldrarna är bärare av genen. Om båda blivande föräldrarna har denna gen är sannolikheten att få ett barn med detta syndrom 1 av 4. En person som bara har en gen för syndromet anses vara bärare, men har inte symtom på sjukdomen. Numera är det ännu inte möjligt att avgöra om en person har genen för denna sjukdom.
Om ett barn föds av föräldrar, varav en inte har en sådan gen, är sannolikheten att han ärver syndromet mycket låg, men han kommer definitivt att vara bärare.
Symtom Usher syndrom
Symtom på Usher syndrom inkluderar hörselnedsättning och onormal ansamling av pigmenterade celler i ögonstrukturerna. Patienten utvecklar sedan degeneration av näthinnan, vilket orsakar synförsämring och slutligen synförlust i de allvarligaste fallen.
Sensorineural hörselnedsättning kan vara mild eller fullständig och utvecklas vanligtvis inte från födseln. Retinapigmentsjukdom kan dock börja utvecklas i barndomen eller senare. Testresultat har visat att central synskärpa kan bibehållas i många år, även när perifert synförsämras (ett tillstånd som kallas "tunnelsyn").
Dessa är sjukdomens huvudsakliga manifestationer, vilka ibland kan kompletteras av andra störningar, såsom psykos och andra psykiska störningar, problem med innerörat och/eller grå starr.
Formulär
Under forskningen identifierades tre typer av denna sjukdom, samt en fjärde form, som är ganska sällsynt.
Typ I av sjukdomen kännetecknas av medfödd fullständig dövhet, såväl som balansstörning. Ofta börjar sådana barn gå först vid 1,5 års ålder. Synförsämring börjar vanligtvis vid 10 års ålder, och den slutliga utvecklingen av tillståndet nattblindhet börjar vid 20 års ålder. Barn med denna typ av sjukdom kan utveckla en progressiv försämring av perifert synfält.
Vid typ II-sjukdom observeras måttlig eller medfödd dövhet. I detta fall upphör ofta inte längre någon försämring av partiell dövhet. Pigmentär retinit börjar utvecklas runt slutet av tonåren eller efter 20 år. Utvecklingen av nattblindhet börjar vanligtvis vid 29-31 års ålder. Synnedsättning vid typ II-patologi fortskrider generellt något långsammare än vid typ I.
Typ III av sjukdomen kännetecknas av progressiv hörselnedsättning, vanligtvis med början under puberteten, samt gradvis utveckling under samma period (något senare än hörselnedsättningen) av retinitis pigmentosa, vilket kan bli en faktor i utvecklingen av progressiv blindhet.
Manifestationer av typ IV-patologi förekommer huvudsakligen hos män. I detta fall observeras även progressiva störningar och förlust av hörsel och syn. Denna form är mycket sällsynt och har vanligtvis en X-kromosomal natur.
Diagnostik Usher syndrom
Diagnos av Usher syndrom ställs baserat på patientens observerade kombination av plötslig dövhet och progressiv synförlust.
Tester
Ett särskilt genetiskt test kan beställas för att upptäcka mutationen.
Elva genetiska loci har hittats som kan orsaka utvecklingen av Usher syndrom, och nio gener har identifierats som definitivt är orsaken till sjukdomen:
- Typ 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Typ 2: ush2a, VLGR1, WHRN.
- Usher syndrom typ 3: USH3A.
NIDCD-forskare har, tillsammans med kollegor från universitet i New York och Israel, identifierat en mutation som heter R245X i Pcdh15-genen som står för en stor andel av typ 1 Usher syndrom i den judiska befolkningen.
För att ta reda på mer om laboratorier som utför kliniska prövningar, besök https://www.genetests.org och sök i laboratoriekatalogen efter "Ushers syndrom".
För att läsa mer om befintliga kliniska prövningar som inkluderar genetisk testning för Usher syndrom, besök https://www.clinicaltrials.gov och sök efter "Usher syndrome" eller "Usher syndrome genetic testing".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentell diagnostik
Det finns flera metoder för instrumentell diagnostik:
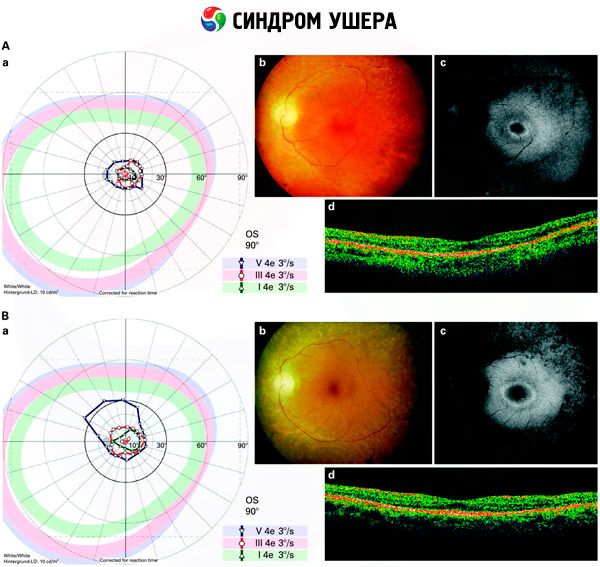
- Undersökning av fundus för att upptäcka förekomsten av pigmentfläckar på näthinnan, samt förträngning av näthinnans kärl;
- Elektroretinogram, vilket möjliggör upptäckt av initiala degenerativa avvikelser i ögats näthinna. Det visar utsläckning av elektroradiografiska banor;
- Ett elektronystagmogram (ENG) mäter ofrivilliga ögonrörelser som kan indikera förekomsten av en obalans.
- Audiometri, som används för att fastställa förekomsten av dövhet och dess svårighetsgrad.
Differentiell diagnos
Usher syndrom måste differentieras från vissa liknande sjukdomar.
Hallgrens syndrom, som kännetecknas av medfödd hörselnedsättning och progressiv synförlust (katarakt och nystagmus utvecklas också). Ytterligare symtom inkluderar ataxi, psykomotoriska störningar, psykos och utvecklingsstörning.
Alströms syndrom, en ärftlig sjukdom där näthinnan degenererar, vilket leder till förlust av central syn. Detta syndrom är förknippat med barnfetma. Samtidigt börjar diabetes mellitus och hörselnedsättning utvecklas efter 10 år.
Röda hund hos en gravid kvinna under första trimestern kan orsaka olika avvikelser i barnets utveckling. Bland konsekvenserna av en sådan avvikelse finns hörselnedsättning, såväl som (eller) problem med synen, och utöver detta olika utvecklingsdefekter.
Vem ska du kontakta?
Behandling Usher syndrom
Det finns för närvarande inget botemedel mot Usher syndrom. Därför består behandlingen i detta fall huvudsakligen av att bromsa processen med synförlust, samt att kompensera för hörselnedsättning. Möjliga behandlingsmetoder inkluderar:
- Intag av vitamin A (vissa ögonläkare tror att höga doser av vitamin A-palmitat kan bromsa, men inte stoppa, utvecklingen av retinit pigmentosa);
- Implantation av speciella elektroniska apparater i patientens öron (hörapparater, cochleaimplantat).
Oftalmologer rekommenderar att de flesta vuxna med vanliga former av retinit pigmentosa tar 15 000 IE (internationella enheter) vitamin A-palmitat dagligen under överinseende. Eftersom personer med typ 1 Usher syndrom inte inkluderades i studien rekommenderas inte höga doser vitamin A för denna patientgrupp. Personer som överväger att ta vitamin A bör diskutera detta behandlingsalternativ med sin läkare. Andra rekommendationer för detta behandlingsalternativ inkluderar:
- Ändra din kost för att inkludera livsmedel med högt innehåll av vitamin A.
- Kvinnor som planerar att bli gravida bör sluta ta höga doser av vitamin A tre månader innan de planerar att bli gravida på grund av en ökad risk för fosterskador.
- Kvinnor som är gravida bör sluta ta höga doser av vitamin A på grund av ökad risk för fosterskador.
Det är också viktigt att anpassa ett sådant barn till det sociala livet. Detta kräver hjälp av specialpedagoger och psykologer. Om patienten har börjat uppleva progressiv synförlust bör hen lära sig att använda teckenspråk.
Prognos
Usher syndrom har en ogynnsam prognos. Synfältet och dess skärpa börjar försämras inom 20-30 års period hos de flesta patienter med denna sjukdom av någon typ. I vissa fall uppstår fullständig bilateral synförlust. Hörselnedsättning, som alltid åtföljs av stumhet, utvecklas mycket snabbt till fullständig bilateral hörselnedsättning.