
Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
Huntingtons sjukdom
Medicinsk expert av artikeln

Senast recenserade: 05.07.2025
Huntingtons sjukdom är en autosomalt dominant neurodegenerativ sjukdom som kännetecknas av progressiv kognitiv nedgång, ofrivilliga rörelser och nedsatt motorisk koordination som börjar i medelåldern. Diagnosen bekräftas genom genetisk testning. Behandlingen är främst symptomatisk. Genetisk testning kan rekommenderas för blodsläktingar. George Huntington beskrev först tillståndet 1872, efter att ha studerat ett familjefall hos invånare på Long Island.
Förekomsten av Huntingtons sjukdom är cirka 10 fall per 100 000 invånare, och med tanke på dess sena debut har cirka 30 personer av 100 000 en 50 % risk att utveckla den under sin livstid. Även om sjukdomen oftast uppträder mellan 35 och 40 år är åldersspannet för debut ganska brett, med tidigast debut vid 3 års ålder och senast vid 90 års ålder. Även om sjukdomen ursprungligen troddes ha 100 % penetrans, tros detta nu inte alltid vara fallet. Hos individer som ärvt genen för sjukdomen från sin far manifesterar sig sjukdomen i genomsnitt 3 år tidigare än hos dem som ärvde den patologiska genen från sin mor. Hos cirka 80 % av patienterna som ärvde den patologiska genen från sin far manifesterar sig sjukdomen före 20 års ålder. Fenomenet med tidigare manifestation av en genetisk defekt hos avkomman kallas anticipation.
 [ 1 ]
[ 1 ]
Vad orsakar Huntingtons sjukdom?
Huntingtons sjukdom har ingen könspreferens. Atrofi av nucleus caudatus påvisas, där små neuroner degenererar och nivån av neurotransmittorer - gamma-aminosmörsyra (GABA) och substans P - sjunker.
En mutantgen med ett ökat antal ("expansion") av CAG (cystein-alanin-glycin) DNA-sekvenser som kodar för aminosyran glutamin är ansvarig för utvecklingen av Huntingtons sjukdom. Produkten av denna gen, det stora proteinet huntingtin, innehåller en alltför stor mängd polyglutaminrester, vilket leder till sjukdomen genom en okänd mekanism. Ju fler CAG-upprepningar, desto tidigare debuterar sjukdomen och desto svårare blir dess förlopp. Från generation till generation kan antalet upprepningar öka, vilket med tiden leder till en försämring av familjens fenotyp.
Trots ett stort intresse för de genetiska och biokemiska förändringarna vid Parkinsons sjukdom var sökandet efter en gen för sjukdomen misslyckat fram till slutet av 1970-talet. Vid den tiden organiserade Nancy Wexler och Allan Tobin en workshop sponsrad av Hereditary Disease Foundation för att diskutera en strategi för att hitta en gen för Huntingtons sjukdom. David Housman, David Botstein och Ray White, som deltog i mötet, föreslog att nyligen utvecklade rekombinanta DNA-tekniker skulle kunna bidra till att uppnå detta mål. En viktig uppgift i projektet var att hitta en stor familj med många generationer av Huntingtons sjukdom för att få DNA-prover. År 1979 lanserades ett gemensamt projekt mellan forskare från Venezuela och USA för att undersöka en stor familj med Huntingtons sjukdom som bodde vid stranden av sjön Maracheibosjön (Venezuela). År 1983 lokaliserades Huntingtons sjukdomsgenen i änden av den korta armen på kromosom 4 (Gusella et al., 1983), och ett decennium senare avslöjades det att mutationen av denna gen består av en ökning av antalet upprepningar av cytosin-adenin-guanin (CAG) trinukleotiden (Huntington's Disease Collaborative Research Group, 1993). Den metod som utvecklats av denna vetenskapliga grupp anses för närvarande vara standard för positionell kloning av nya gener.
Medan vildtypsgenen har en sträcka på 10–28 CAG-upprepningar, har den muterade formen av genen som orsakar Huntingtons sjukdom en ökad sträcka från 39 till mer än 100 CAG-upprepningar. Upptäckten av expansionen av trinukleotidupprepningar har bidragit till att förklara många av sjukdomens kliniska egenskaper. I synnerhet hittades en omvänd korrelation mellan debutåldern och längden på regionen med upprepade trinukleotider. Förväntan på faderns nedärvning kan förklaras av det faktum att en ökning av antalet upprepningar ofta sker hos män under spermatogenesen. Analys av nya mutationer har visat att de vanligtvis inträffar när en av föräldrarna, vanligtvis fadern, hade ett CAG-upprepningsantal högre än 28; i detta fall ökade antalet av dessa upprepningar i nästa generation. Det har nu fastställts att om antalet upprepningar inte är mer än 28, överförs det stabilt från generation till generation. Om antalet upprepningar är från 29 till 35 uppträder inga symtom på Huntingtons sjukdom, men vid överföring till avkomman kan längden på denna region öka. Om antalet upprepningar är från 36 till 39 kan sjukdomen i vissa fall (men inte alltid) manifestera sig kliniskt (ofullständig penetrans), och vid överföring till avkomman är en ökning av antalet trinukleotidupprepningar möjlig. Om antalet upprepningar överstiger 40 uppstår sjukdomen i nästan alla fall, och vid överföring till avkomman är ytterligare expansion av upprepningar möjlig. Orsakerna till ökningen av antalet upprepningar förblir okända.
Patomorfologin för Huntingtons sjukdom
Huntingtons sjukdom kännetecknas av neuronal förlust främst i nucleus caudatus och putamen, och i viss mån även i cortex och andra hjärnstrukturer. Den totala hjärnvikten vid Huntingtons sjukdom minskar inte bara av en minskning av antalet neuroner, utan också av förlusten av vit substans. I hjärnbarken är celler i lager V och VI mest drabbade. Svårighetsgraden av mikro- och makroskopiska degenerativa förändringar (justerade för ålder vid dödsfall) korrelerar med antalet CAG-upprepningar. Detaljerad patologisk analys av förändringar i flera hundra fall av Huntingtons sjukdom har visat att degeneration av striatum börjar i den dorsomediala delen av nucleus caudatus och den dorsolaterala delen av putamen, och sedan sprider sig ventralt. Olika grupper av neuroner i nucleus caudatus och putamen påverkas i olika grad. Interneuroner i striatum förblir relativt intakta, men vissa projektionsneuroner påverkas selektivt. Vid den juvenila formen av Huntingtons sjukdom är patomorfologiska förändringar i striatum mer uttalade och mer utbredda och involverar hjärnbarken, lillhjärnan, talamus och globus pallidus.
Neurokemiska förändringar vid Huntingtons sjukdom
GABA. Neurokemiska studier av hjärnan hos patienter med Huntingtons sjukdom visade en signifikant minskning av GABA-koncentrationen i striatum. Efterföljande studier bekräftade att Huntingtons sjukdom är associerad med en minskning av antalet GABAerga neuroner och visade att GABA-koncentrationerna är reducerade inte bara i striatum utan även i dess projektionszoner - de externa och interna segmenten av globus pallidus och substantia nigra. I hjärnan vid Huntingtons sjukdom detekterades även förändringar i GABA-receptorer med hjälp av receptorbindningsstudier och in situ-hybridisering av mRNA. Antalet GABA-receptorer var måttligt reducerat i nucleus caudatus och putamen, men ökat i den retikulära delen av substantia nigra och det externa segmentet av globus pallidus, vilket sannolikt beror på denervationsöverkänslighet.
Acetylkolin. Acetylkolin används som en neurotransmittor av stora icke-taggiga interneuroner i striatum. Tidiga obduktionsstudier på patienter med Huntingtons sjukdom visade minskad kolinacetyltransferas (ChAT)-aktivitet i striatum, vilket tyder på en förlust av kolinerga neuroner. Jämfört med den signifikanta minskningen av GABAerga neuroner är dock kolinerga interneuroner relativt skonade. Därför är densiteten av acetylkolinesteraspositiva neuroner och ChAT-aktivitet i striatum faktiskt relativt förhöjd jämfört med åldersmatchade kontroller.
Substans P. Substans P finns i många medelstora taggiga neuroner i striatum, vilka huvudsakligen skjuter ut till det interna segmentet av globus pallidus och substantia nigra och vanligtvis även innehåller dynorfin och GABA. Substans P-nivåerna i striatum och pars reticularis i substantia nigra är reducerade vid Huntingtons sjukdom. I sjukdomens terminala stadium har immunhistokemiska studier visat en signifikant minskning av antalet neuroner som innehåller substans P. I tidigare stadier är neuroner som innehåller substans P och skjuter ut till det interna segmentet av globus pallidus relativt skonade, jämfört med neuroner som skjuter ut till pars reticularis i substantia nigra.
Opioidpeptider. Enkefalin finns i de medeltunga taggiga projektionerna av GABAerga neuroner i den indirekta signalvägen, vilka projicerar till det externa segmentet av globus pallidus och bär D2-receptorer. Immunhistokemiska studier har visat att enkefalininnehållande neuroner som projicerar till det externa segmentet av globus pallidus förloras tidigt vid Huntingtons sjukdom. Dessa celler dör tydligen tidigare än substans P-innehållande celler som projicerar till det interna segmentet av globus pallidus.
Katekolaminer. Neuroner som innehåller biogena aminer (dopamin, serotonin) och som utskjuter mot striatum finns i den kompakta delen av substantia nigra, ventrala tegmentum och raphe-kärnorna. Medan noradrenerga utsprång mot det mänskliga striatum är minimala, är serotonin- och dopaminnivåerna (per gram vävnad) i striatum förhöjda, vilket indikerar att dessa afferenta utsprång bevaras trots den markanta förlusten av striatums egna neuroner. Dopaminerga neuroner i substantia nigra förblir intakta i både klassiska och juvenila former av Huntingtons sjukdom.
Somatostatin/neuropeptid Y och kväveoxidsyntetas. Mätning av somatostatin- och neuropeptid Y-nivåer i striatum vid Huntingtons sjukdom visade en 4-5-faldig ökning jämfört med normala vävnader. Immunhistokemiska studier visade absolut bevarande av striatala interneuroner innehållande neuropeptid Y, somatostatin och kväveoxidsyntetas. Således är dessa neuroner resistenta mot den patologiska processen.
Exciterande aminosyror. Det har föreslagits att selektiv celldöd vid Huntingtons sjukdom beror på en glutamatinducerad neurotoxisk effekt. Nivåerna av glutamat och kinolinsyra (ett endogent neurotoxin som är en biprodukt av serotoninmetabolism och en agonist av glutamatreceptorer) i striatum vid Huntingtons sjukdom är något förändrade, men en nyligen genomförd studie med MR-spektroskopi visade en ökning av glutamatnivåerna in vivo. Nivån av det glialenzym som ansvarar för syntesen av kinolinsyra i striatum vid Huntingtons sjukdom är cirka 5 gånger högre jämfört med normalt, medan aktiviteten hos det enzym som säkerställer nedbrytningen av kinolinsyra endast är 20–50 % högre vid Huntingtons sjukdom. Således kan syntesen av kinolinsyra vara ökad vid Huntingtons sjukdom.
Studier av exciterande aminosyrareceptorer (EAA) vid Huntingtons sjukdom har visat en signifikant minskning av antalet NMDA-, AMPA-, kainat- och metabotropa glutamatreceptorer i striatum, samt AMPA- och kainatreceptorer i hjärnbarken. I det sena stadiet av Huntingtons sjukdom var NMDA-receptorer praktiskt taget frånvarande, medan en signifikant minskning av antalet av dessa receptorer noterades i de prekliniska och tidiga stadierna.
Selektiv känslighet. Vid Huntingtons sjukdom förloras vissa typer av striatala celler selektivt. De medelstora taggiga neuronerna, som skjuter ut till det externa segmentet av globus pallidus och innehåller GABA och enkefalin, dör mycket tidigt i sjukdomen, liksom de neuroner som innehåller GABA och substans P och skjuter ut till den retikulära delen av substantia nigra. Förlusten av neuroner som innehåller GABA och enkefalin och skjuter ut till det externa segmentet av globus pallidus avhämmar denna struktur, vilket i sin tur leder till aktiv hämning av subthalamuskärnan. Den minskade aktiviteten i subthalamuskärnan kan tydligen förklara de koreformiga rörelserna som uppstår vid Huntingtons sjukdom. Det har länge varit känt att fokala lesioner i subthalamuskärnan kan orsaka korea. Förlust av GABA- och substans P-neuroner som skjuter ut till substantia nigra pars reticularis är sannolikt ansvarig för de okulomotoriska störningar som ses vid Huntingtons sjukdom. Denna signalväg hämmar normalt substantia nigra pars reticularis-neuroner som projicerar till superior colliculus, vilka i sin tur reglerar sackader. Vid juvenil Huntingtons sjukdom påverkas de ovan nämnda signalvägarna hårdare och dessutom förloras striatala projektioner till det interna segmentet av globus pallidus tidigt.
Proteinet huntingtin, som kodas av genen vars mutation orsakar Huntingtons sjukdom, finns i olika strukturer i hjärnan och andra vävnader. Huntingtin finns normalt huvudsakligen i neuronernas cytoplasma. Proteinet finns i de flesta neuroner i hjärnan, men nya data visar att dess innehåll är högre i matrixneuroner än i striosomala neuroner, och högre i projektionsneuroner än i interneuroner. Således korrelerar neuronernas selektiva känslighet med deras huntingtininnehåll, vilket normalt finns i vissa neuronala populationer.
Precis som i hjärnan hos patienter med Huntingtons sjukdom bildar huntingtin täta aggregat i neuronkärnan hos möss som är transgena för det N-terminala fragmentet av Huntingtons sjukdomsgen med ett utökat antal upprepningar. Dessa intranukleära inneslutningar bildas i striatala projektionsneuroner (men inte i interneuroner). Hos transgena möss bildas inneslutningarna flera veckor innan symtomen börjar. Dessa data tyder på att huntingtinprotein som innehåller ett ökat antal glutaminrester vars inneslutningar kodar för trinukleotidupprepningar, eller ett fragment av det, ackumuleras i kärnan och följaktligen kan försämra dess kontroll över cellulära funktioner.
Symtom på Huntingtons sjukdom
Åldern då de första symtomen uppträdde hos patienter med Huntingtons sjukdom är svår att fastställa med precision, eftersom sjukdomen manifesterar sig gradvis. Förändringar i personlighet och beteende, milda koordinationsstörningar kan uppstå många år innan mer tydliga symtom uppstår. När diagnosen ställs har de flesta patienter koreiska rörelser, nedsatt koordination av fina rörelser och långsam generering av frivilliga sackader. Allt eftersom sjukdomen fortskrider försämras förmågan att organisera sina aktiviteter, minnet minskar, talet blir svårt, ögonmotoriska störningar och nedsatt prestation av koordinerade rörelser ökar. Även om det i ett tidigt skede av sjukdomen inte sker några förändringar i muskler och hållning, kan dystoniska ställningar utvecklas allt eftersom den fortskrider, vilket med tiden kan bli ett dominerande symptom. I ett sent skede blir talet sluddrigt, det blir betydligt svårt att svälja, det blir omöjligt att gå. Huntingtons sjukdom fortskrider vanligtvis under 15-20 år. I det terminala skedet är patienten hjälplös och kräver ständig vård. Den dödliga utgången är inte direkt relaterad till den primära sjukdomen, utan till dess komplikationer, till exempel lunginflammation.
Demens vid Huntingtons sjukdom
ICD-10-kod
P02.2. Demens vid Huntingtons sjukdom (G10).
Demens utvecklas som en av manifestationerna av en systemisk degenerativ-atrofisk process med övervägande skador på hjärnans striatala system och andra subcoekala kärnor. Den ärvs autosomalt dominant.
Som regel manifesterar sig sjukdomen under det tredje eller fjärde decenniet av livet med koreoform hyperkinesi (särskilt i ansikte, armar, axlar, gång), personlighetsförändringar (upphetsade, hysteriska och schizoida typer av personlighetsavvikelser), psykotiska störningar (speciell depression med dysterhet, surhet, dysfori; paranoid sinnesstämning).
Av särskild betydelse för diagnostiken är kombinationen av koreoform hyperkinesi, demens och ärftlig belastning. Följande är specifikt för denna demens:
- långsam progression (i genomsnitt 10–15 år): dissociation mellan den återstående förmågan att ta hand om sig själv och uppenbar intellektuell inkompetens i situationer som kräver produktivt mentalt arbete (konceptuellt tänkande, inlärning av nya saker);
- uttalad ojämnhet i mental prestation, som är baserad på grova uppmärksamhetsstörningar och ostadighet i patientens attityder ("ryckigt" tänkande, liknande hyperkinesi);
- atypikalitet av uppenbara kränkningar av högre kortikala funktioner;
- omvänt samband mellan ökningen av demens och svårighetsgraden av psykotiska störningar.
Med hänsyn till den höga andelen psykotiska (paranoida vanföreställningar om svartsjuka, förföljelse) och dysforiska störningar i sjukdomens kliniska bild, utförs behandlingen med olika neuroleptika som blockerar dopaminerga receptorer (fentiazin- och butyrofenonderivat) eller minskar nivån av dopamin i vävnader (reserpin).
Haloperidol (2–20 mg/dag), tiaprid (100–600 mg/dag) i högst tre månader, tioridazin (upp till 100 mg/dag), reserpin (0,25–2 mg/dag) och det antikonvulsiva läkemedlet klonazepam (1,5–6 mg/dag) används. Dessa läkemedel hjälper till att minska hyperkinesi, jämna ut affektiv spänning och kompensera för personlighetsstörningar.
Behandling av psykiska störningar i öppenvården utförs med hänsyn till patientens ledande syndrom, ålder och allmäntillstånd. Vid öppenvård är behandlingsprinciperna desamma (kontinuerlig underhållsbehandling av rörelsestörningar, periodiskt läkemedelsbyte). Lägre doser av neuroleptika används vid öppenvård.
Rehabiliteringsåtgärder för mild och måttlig demens inkluderar arbetsterapi, psykoterapi och kognitiv träning. Det är nödvändigt att samarbeta med familjemedlemmar och ge psykologiskt stöd till personer som vårdar patienten. Den huvudsakliga metoden för att förebygga sjukdom är medicinsk och genetisk rådgivning av patientens närmaste anhöriga med remiss för DNA-analys vid beslut om barnafödande.
Prognosen är generellt ogynnsam. Sjukdomsförloppet är långsamt progressivt och sjukdomen leder vanligtvis till döden efter 10–15 år.
[ 18 ]
Vad stör dig?
Diagnos av Huntingtons sjukdom
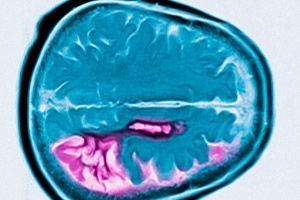
Diagnosen baseras på typiska symtom, familjehistoria och genetiska tester. På grund av atrofi av nucleus caudatus huvud visar MR och cerebrospinalkameraundersökningar förstoring av hjärnventriklarna i sjukdomens sena skede.
Behandling av Huntingtons sjukdom
Behandling av Huntingtons sjukdom är symptomatisk. Korea och agitation kan delvis dämpas med neuroleptika (t.ex. klorpromazin 25-300 mg oralt 3 gånger dagligen, haloperidol 5-45 mg oralt 2 gånger dagligen) eller reserpin 0,1 mg oralt en gång dagligen. Doserna ökas till maximalt tolererbart värde (innan biverkningar uppstår, såsom dåsighet, parkinsonism; för reserpin, hypotoni). Målet med empirisk behandling är att minska glutamaterg transmission via Nmetyl-O-aspartatreceptorer och upprätthålla energiproduktionen i mitokondrierna. Behandling som syftar till att öka GABA i hjärnan är ineffektiv.
Genetiska tester och rådgivning är viktiga eftersom symtom på sjukdomen uppträder efter fertil ålder. Personer med positiv familjehistoria och de som är intresserade av testning hänvisas till specialiserade centra, med hänsyn till alla etiska och psykologiska implikationer.
Symtomatisk behandling av Huntingtons sjukdom
Det finns ingen effektiv behandling som kan stoppa utvecklingen av Huntingtons sjukdom. Flera studier av olika läkemedel har genomförts, men ingen signifikant effekt har uppnåtts. Neuroleptika och andra dopaminreceptorantagonister används i stor utsträckning för att korrigera psykiska störningar och ofrivilliga rörelser hos patienter med Huntingtons sjukdom. Ofrivilliga rörelser återspeglar en obalans mellan de dopaminerga och GABAerga systemen. Följaktligen används neuroleptika för att minska överdriven dopaminerg aktivitet. Dessa läkemedel kan dock i sig orsaka betydande kognitiva och extrapyramidala biverkningar. Dessutom, förutom i de fall där patienten utvecklar psykos eller agitation, har deras effektivitet inte bevisats. Neuroleptika orsakar eller förvärrar ofta dysfagi eller andra rörelsestörningar. Nyare generationers neuroleptika som risperidon, klozapin och olanzapin kan vara särskilt användbara vid behandling av Huntingtons sjukdom eftersom de orsakar färre extrapyramidala biverkningar men kan minska paranoida symtom eller ökad irritabilitet.
Tetrabenazin och reserpin minskar också aktiviteten i det dopaminerga systemet och kan minska svårighetsgraden av ofrivilliga rörelser i sjukdomens tidiga stadier. Dessa läkemedel kan dock orsaka depression. Eftersom själva sjukdomen ofta orsakar depression begränsar denna biverkning användningen av reserpin och tetrabenazin avsevärt. I sjukdomens sena stadier dör celler som bär dopaminreceptorer, vilket försvagar eller förlorar dopaminreceptorantagonisternas effektivitet.
Neuroleptika, antidepressiva och ångestdämpande medel används för att behandla psykos, depression och irritabilitet hos patienter med Huntingtons sjukdom, men de bör endast förskrivas så länge patienten faktiskt har dessa symtom. Läkemedel som kan vara till hjälp i ett skede av sjukdomen kan bli ineffektiva eller till och med skadliga allt eftersom sjukdomen fortskrider.
GABA-receptoragonister har testats på patienter med Huntingtons sjukdom, eftersom Huntingtons sjukdom har visat sig ha en signifikant minskning av GABA-nivåerna i striatum, samt överkänslighet hos GABA-receptorer i dess projektionsområden. Bensodiazepiner har visat sig effektiva i fall där ofrivilliga rörelser och kognitiv nedsättning förvärras av stress och ångest. Låga doser av dessa läkemedel bör förskrivas för att undvika oönskade lugnande effekter. Hos de flesta patienter med Huntingtons sjukdom leder inget av läkemedlen till en signifikant förbättring av livskvaliteten.
Vid tidig debut av Huntingtons sjukdom med parkinsonsymtom kan dopaminerga läkemedel prövas, men deras effektivitet är begränsad. Dessutom kan levodopa orsaka eller öka myoklonus hos dessa patienter. Samtidigt kan baklofen minska stelhet hos vissa patienter med Huntingtons sjukdom.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Förebyggande (neuroprotektiv) behandling av Huntingtons sjukdom
Även om den genetiska defekten vid Huntingtons sjukdom är känd, är det fortfarande oklart hur den leder till selektiv neuronal degeneration. Det antas att förebyggande behandlingar som syftar till att minska oxidativ stress och excitotoxicitet potentiellt kan bromsa eller stoppa sjukdomsprogressionen. Situationen kan likna hepatolentikulär degeneration, där den genetiska defekten förblev okänd i många år, men förebyggande behandlingar som syftar till den sekundära effekten, kopparackumulering, "botades". I detta avseende har hypotesen att Huntingtons sjukdom är förknippad med en störning i energimetabolismen och celldöd på grund av excitotoxicitet väckt särskild uppmärksamhet. Själva sjukdomen kan orsaka celldöd på grund av intranukleär aggregering av N-terminala fragment av huntingtin, vilket stör cellulära och metaboliska funktioner. Denna process kan påverka vissa grupper av neuroner i större utsträckning än andra på grund av deras högre känslighet för excitotoxisk skada. I detta fall kommer förebyggande behandling med exciterande aminosyrareceptorantagonister eller medel som förhindrar fria radikalskador att kunna förhindra eller fördröja sjukdomens uppkomst och progression. I laboratoriemodeller av amyotrofisk lateralskleros har det visats att antioxidanter och receptorantagonister (RAA) kan bromsa sjukdomsprogressionen. Liknande metoder kan vara effektiva vid Huntingtons sjukdom. Kliniska prövningar av glutamatreceptorantagonister och medel som förstärker funktionen av komplex II i den mitokondriella elektrontransportkedjan pågår för närvarande.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]